Bệnh viện đa khoa tỉnh Hà Nam được thành lập năm 1954, là bệnh viện hạng I Quy mô 1270 giường bệnh. Bệnh viện được sự đầu tư của nhà nước, Bộ Y tế, tỉnh Hà Nam và đặc biệt sự đóng góp, nỗ lực của tập thể cán bộ viên chức của bệnh viện qua nhiều thế hệ, trải qua thời gian xây dựng và phát triển đến nay bệnh viện có một cơ sở vật chất hoàn chỉnh cùng với đội ngũ cán bộ y tế vững mạnh về chuyên môn.
Chức năng, nhiệm vụ:
- Cấp cứu, khám chữa bệnh
- Đào tạo cán bộ y tế
- Nghiên cứu khoa học về y học
- Chỉ đạo tuyến dưới về
chuyên môn, kỹ thuật
- Phòng bệnh
- Thực hiện các chương trình mục tiêu Quốc gia
- Quản lý kinh tế trong bệnh viện.


T03
07
Trong 2 ngày 24 và 25/2, tại Nhà thi đấu đa năng tỉnh Hà Nam, Công đoàn ngành Y tế tổ chức Giải t...

T02
28
Nghề Y là một nghề đặc biệt, những người Thầy thuốc tâm huyết, gắn bó với Nghề Y cũng là những ng...

T02
28
Ngân hàng thương mại cổ phần Công thương Việt Nam chi nhánh Hà Nam đã tặng 300 tr...

T06
02
Hội Tin học Y tế Việt Nam, Bệnh viện Đa khoa tỉnh Hà Nam tổ chức hội nghị Bàn giao và khai ...
T03
18
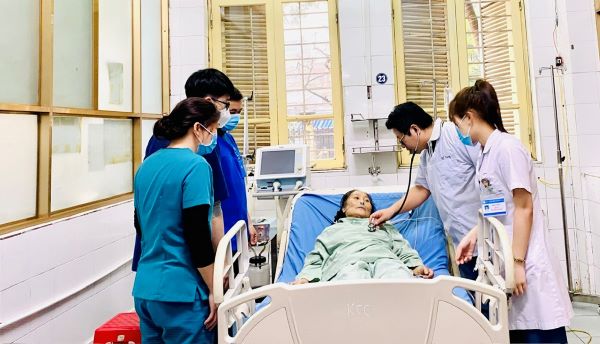
11h 30 phút ngày chủ nhật 10/3/2024, tại khoa Cấp cứu, Bệnh viện Đa khoa tỉnh Hà Nam, các đi...

T03
07
Ngày 5/3/2024, Khoa Sơ sinh, Bệnh viện Đa khoa tỉnh Hà Nam có 2 niềm vui lớn đó là có thêm hai ch...

T03
01
Đoàn công tác của Bệnh viện Bạch Mai do TS.BS. Nghiêm Trung Dũng, Phó Giám đốc Trung tâm Th...

T03
01
Với mong muốn góp phần chia sẻ, khích lệ tinh thần, giúp các em nhỏ chiến thắng bệnh tật và có mộ...
Giao hàng trên toàn quốc
Thành tiền: